
Molecular and Structural Parallels between Gluten Pathogenic Peptides and Bacterial-Derived Proteins by Bioinformatics Analysis


Abstract
:1. Introduction
2. Results and Discussion
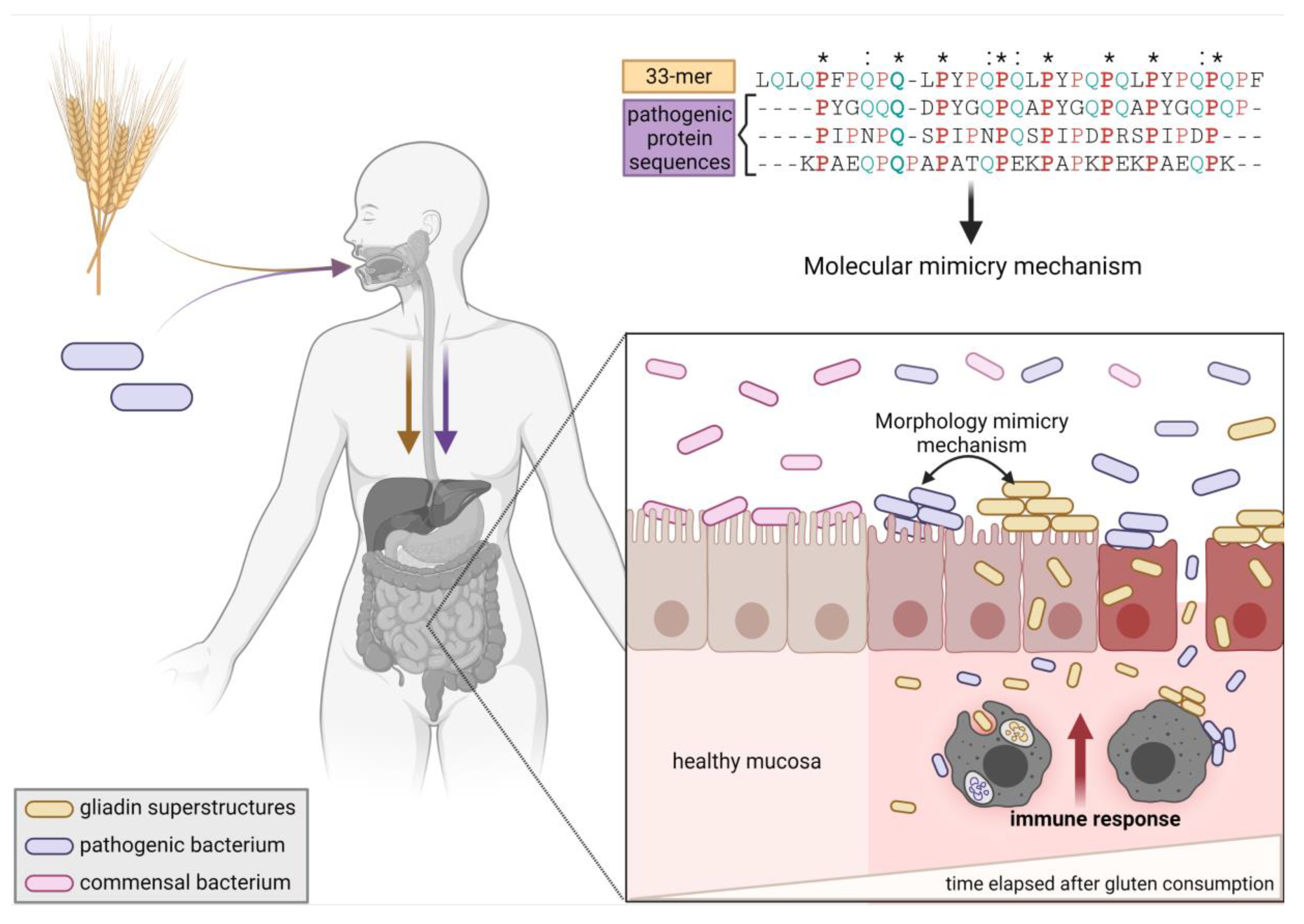
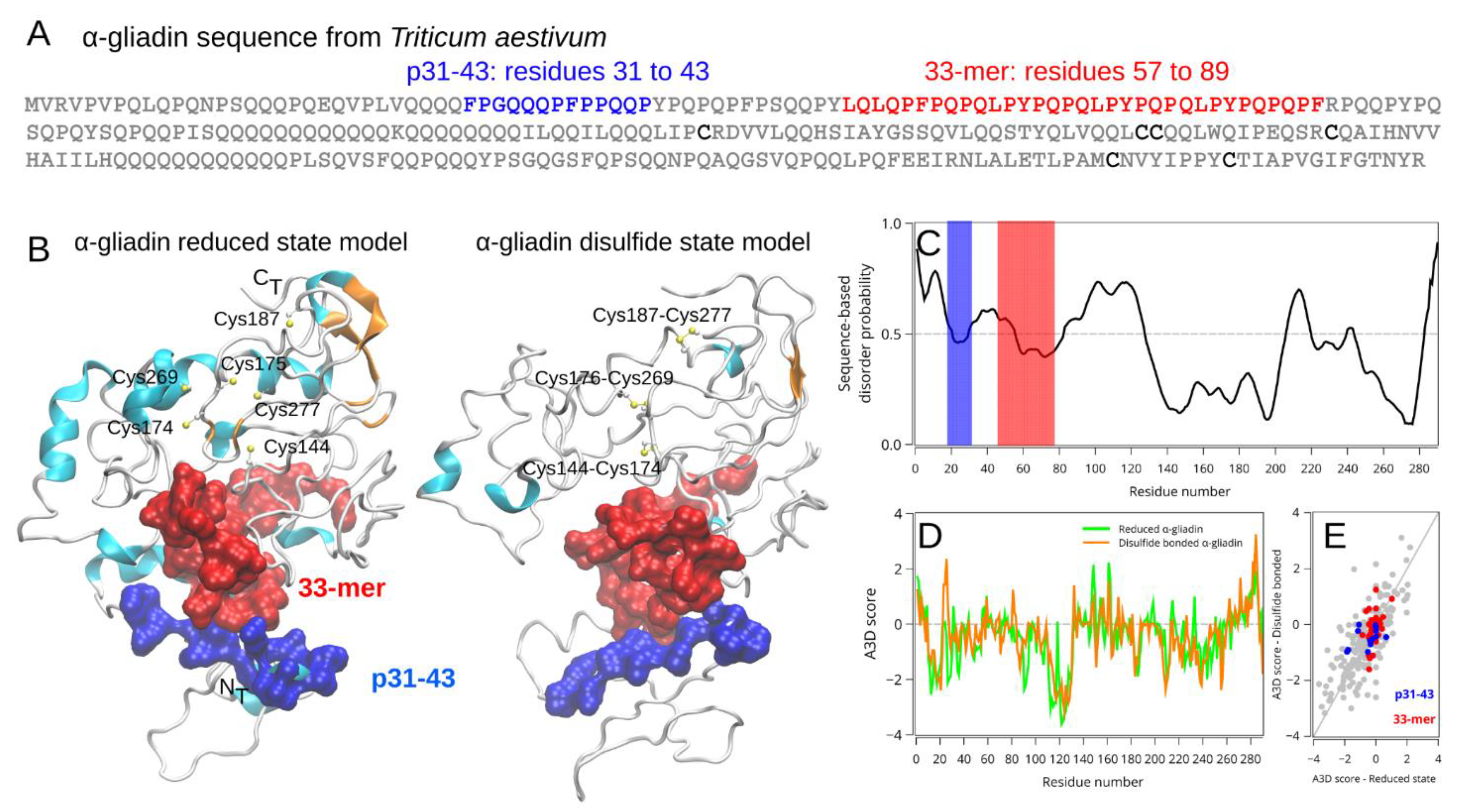
2.1. High Sequence and Structural Similarity of 33-mer and p31-43 Sequences in Pathogen-Derived Proteins
2.2. The Structural Evaluation of Gliadin Peptides Harboring Pathogen-Related Proteins Similarity Regions and Their Function
2.2.1. Spatial Localization and Structural Information of the Relevant Gliadin-Derived Peptides in the α-2-Gliadin 3D Model
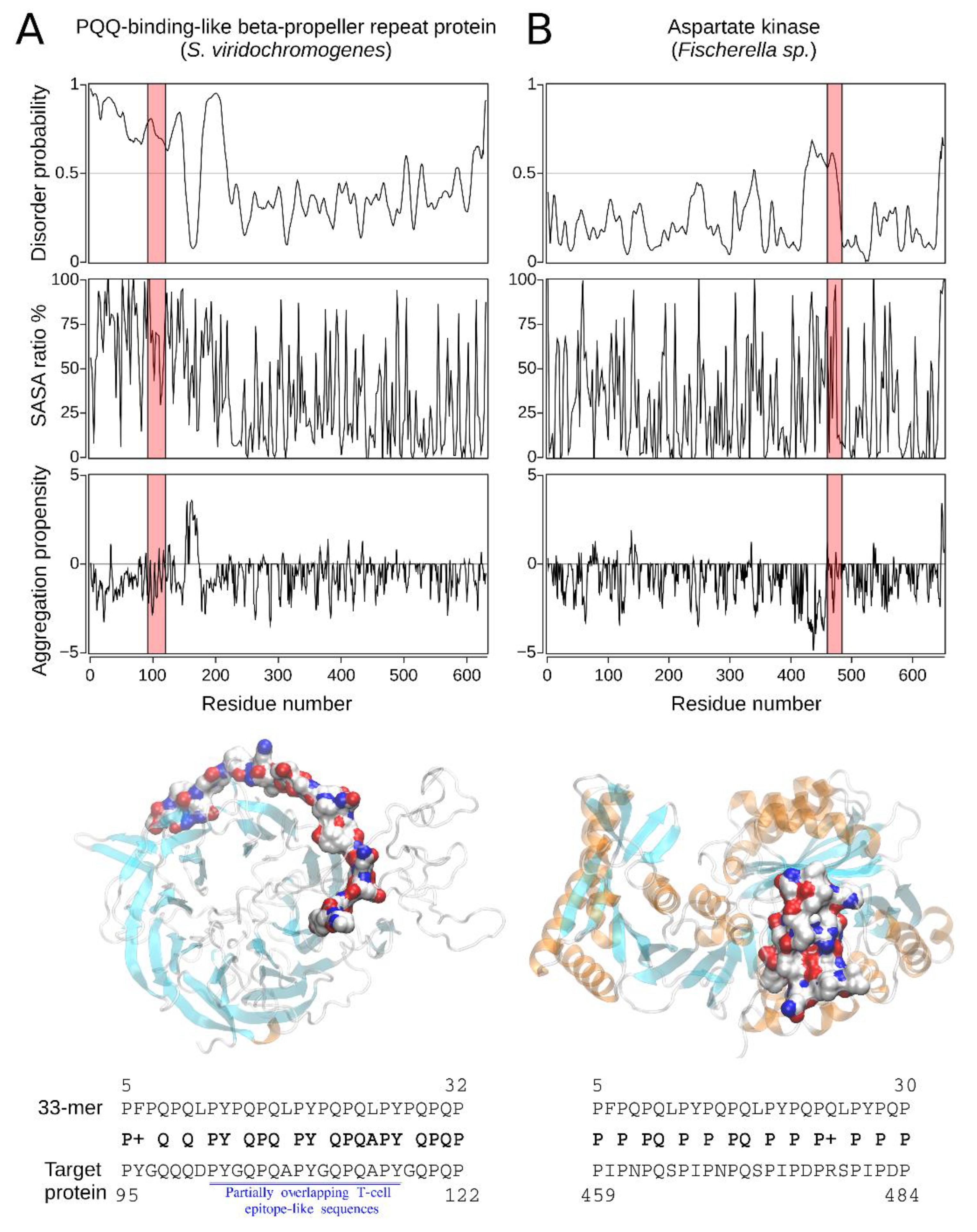
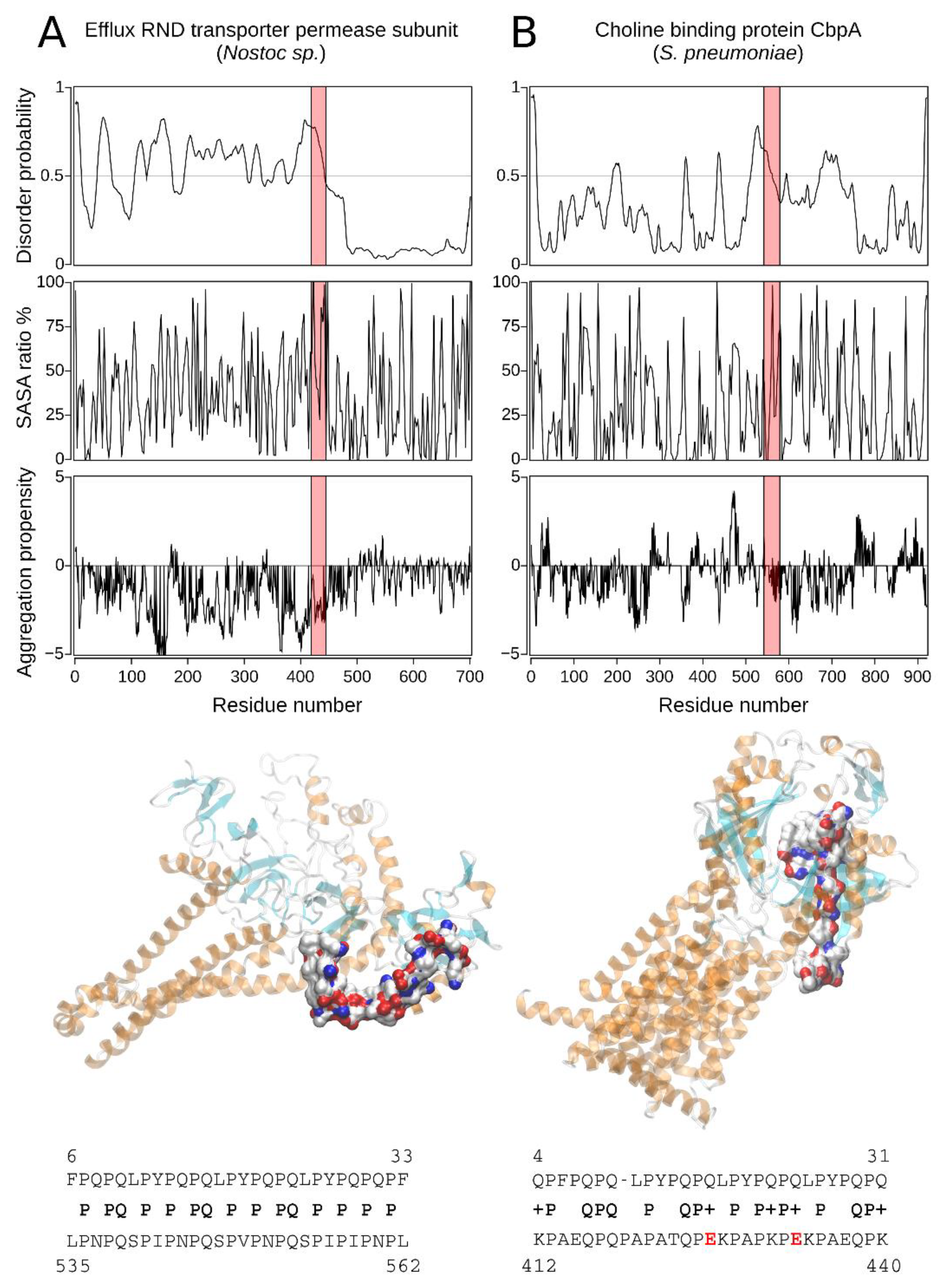
2.2.2. The Homology Models of Pathogen-Related Proteins Containing Sequences Similar to 33-mer Gliadin Peptide and Their Function
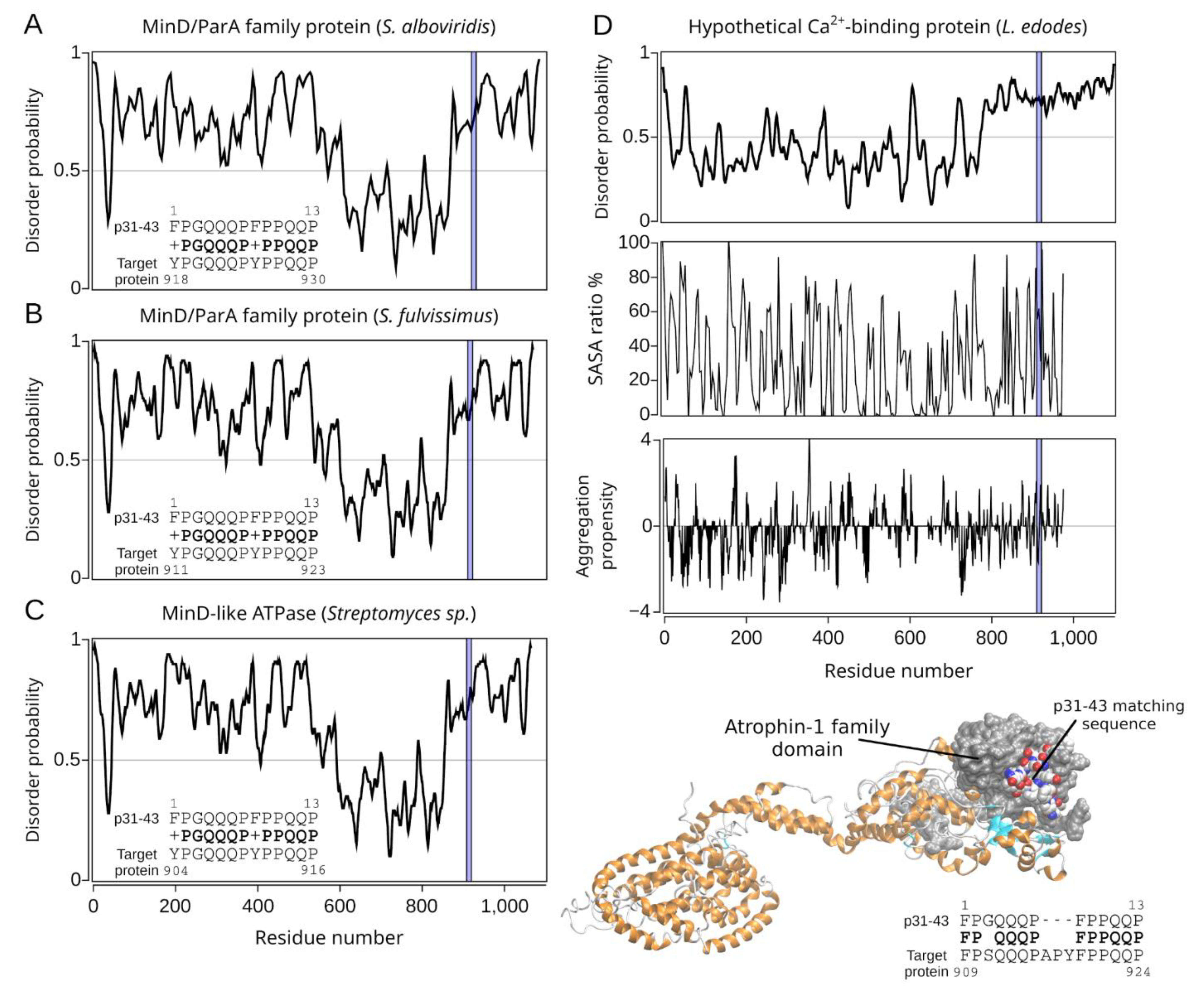
2.2.3. The Homology Models of Pathogen-Related Proteins Containing the p31-43 Similar Sequence and Their Function
2.3. Primary Structure Analysis and Potential Functions
2.3.1. CeD-T-Cell Epitopes
2.3.2. SH3/WW Domains Binders
2.4. Gliadin Peptide Superstructures, Pathogen Morphology, and Dysbiosis as Triggers of the Innate Immune Response: The Hypothesis
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CeD | celiac disease |
| ID | intrinsic disorder |
| MHC | major histocompatibility complex |
| PPII | type-II polyproline |
| SASA | solvent-accessible surface area |
References
- Cabanillas, B. Gluten-related disorders: Celiac disease, wheat allergy, and nonceliac gluten sensitivity. Crit. Rev. Food Sci. Nutr. 2020, 60, 2606–2621. [Google Scholar] [CrossRef]
- Taraghikhah, N.; Ashtari, S.; Asri, N.; Shahbazkhani, B.; Al-Dulaimi, D.; Rostami-Nejad, M.; Rezaei-Tavirani, M.; Razzaghi, M.R.; Zali, M.R. An updated overview of spectrum of gluten-related disorders: Clinical and diagnostic aspects. BMC Gastroenterol. 2020, 20, 258. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Corazza, G.R. Coeliac disease. Lancet 2009, 373, 1480–1493. [Google Scholar] [CrossRef]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural Basis for Gluten Intolerance in Celiac Sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef]
- Petersen, J.; Ciacchi, L.; Tran, M.T.; Loh, K.L.; Kooy-Winkelaar, Y.; Croft, N.P.; Hardy, M.I.; Chen, Z.; McCluskey, J.; Anderson, R.P.; et al. T cell receptor cross-reactivity between gliadin and bacterial peptides in celiac disease. Nat. Struct. Mol. Biol. 2020, 27, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Bethune, M.T.; Khosla, C. Parallels between Pathogens and Gluten Peptides in Celiac Sprue. PLoS Pathog. 2008, 4, e34. [Google Scholar] [CrossRef]
- Verdu, E.F.; Schuppan, D. The enemy within the gut: Bacterial pathogens in celiac autoimmunity. Nat. Struct. Mol. Biol. 2019, 27, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Lammers, K.M.; Herrera, M.G.; Dodero, V.I. Translational Chemistry Meets Gluten-Related Disorders. ChemistryOpen 2018, 7, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Doshi, N.; Mitragotri, S. Macrophages Recognize Size and Shape of Their Targets. PLoS ONE 2010, 5, e10051. [Google Scholar] [CrossRef]
- Swartzwelter, B.; Fux, A.; Johnson, L.; Swart, E.; Hofer, S.; Hofstätter, N.; Geppert, M.; Italiani, P.; Boraschi, D.; Duschl, A.; et al. The Impact of Nanoparticles on Innate Immune Activation by Live Bacteria. Int. J. Mol. Sci. 2020, 21, 9695. [Google Scholar] [CrossRef]
- Herrera, M.G.; Nicoletti, F.; Gras, M.; Dörfler, P.W.; Tonali, N.; Hannappel, Y.; Ennen, I.; Hütten, A.; Hellweg, T.; Lammers, K.M.; et al. Pepsin Digest of Gliadin Forms Spontaneously Amyloid-Like Nanostructures Influencing the Expression of Selected Pro-Inflammatory, Chemoattractant, and Apoptotic Genes in Caco-2 Cells: Implications for Gluten-Related Disorders. Mol. Nutr. Food Res. 2021, 65, 2100200. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Auricchio, S.; Picard, J.; Osman, M.; Quaratino, S.; Londei, M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003, 362, 30–37. [Google Scholar] [CrossRef]
- Londei, M.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Quaratino, S.; Maiuri, L. Gliadin as a stimulator of innate responses in celiac disease. Mol. Immunol. 2005, 42, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.-W.; Bergseng, E.; Molberg, Ø.; Xia, J.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Antigen Presentation to Celiac Lesion-Derived T Cells of a 33-Mer Gliadin Peptide Naturally Formed by Gastrointestinal Digestion. J. Immunol. 2004, 173, 1757–1762. [Google Scholar] [CrossRef]
- Fraser, J.S.; Engel, W.; Ellis, H.J.; Moodie, S.J.; Pollock, E.L.; Wieser, H.; Ciclitira, P.J. Coeliac disease: In vivo toxicity of the putative immunodominant epitope. Gut 2003, 52, 1698–1702. [Google Scholar] [CrossRef]
- Sollid, L.M. Intraepithelial Lymphocytes in Celiac Disease: License to Kill Revealed. Immunity 2004, 21, 303–304. [Google Scholar] [CrossRef]
- Herrera, M.G.; Zamarreño, F.; Costabel, M.; Ritacco, H.; Hütten, A.; Sewald, N.; Dodero, V.I. Circular dichroism and electron microscopy studies in vitro of 33-mer gliadin peptide revealed secondary structure transition and supramolecular organization. Biopolymers 2014, 101, 96–106. [Google Scholar] [CrossRef]
- Herrera, M.G.; Benedini, L.; Lonez, C.; Schilardi, P.L.; Hellweg, T.; Ruysschaert, J.-M.; Dodero, V.I. Self-assembly of 33-mer gliadin peptide oligomers. Soft Matter 2015, 11, 8648–8660. [Google Scholar] [CrossRef]
- Herrera, M.; Vazquez, D.; Sreij, R.; Drechsler, M.; Hertle, Y.; Hellweg, T.; Dodero, V. Insights into gliadin supramolecular organization at digestive pH 3.0. Colloids Surf. B Biointerfaces 2018, 165, 363–370. [Google Scholar] [CrossRef]
- Herrera, M.G.; Veuthey, T.V.; Dodero, V.I. Self-organization of gliadin in aqueous media under physiological digestive pHs. Colloids Surf. B Biointerfaces 2016, 141, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Herrera, M.G.; Pizzuto, M.; Lonez, C.; Rott, K.; Hütten, A.; Sewald, N.; Ruysschaert, J.-M.; Dodero, V.I. Large supramolecular structures of 33-mer gliadin peptide activate toll-like receptors in macrophages. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1417–1427. [Google Scholar] [CrossRef]
- Amundarain, M.J.; Herrera, M.G.; Zamarreño, F.; Viso, J.F.; Costabel, M.D.; Dodero, V.I. Molecular mechanisms of 33-mer gliadin peptide oligomerisation. Phys. Chem. Chem. Phys. 2019, 21, 22539–22552. [Google Scholar] [CrossRef] [PubMed]
- Falcigno, L.; Calvanese, L.; Conte, M.; Nanayakkara, M.; Barone, M.V.; D’Auria, G. Structural Perspective of Gliadin Peptides Active in Celiac Disease. Int. J. Mol. Sci. 2020, 21, 9301. [Google Scholar] [CrossRef]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Auricchio, R.; De Musis, C.; Discepolo, V.; Miele, E.; Jabri, B.; Troncone, R.; Auricchio, S.; et al. P31–43, an undigested gliadin peptide, mimics and enhances the innate immune response to viruses and interferes with endocytic trafficking: A role in celiac disease. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Herrera, M.G.; Castro, M.F.G.; Prieto, E.; Barrera, E.; Dodero, V.I.; Pantano, S.; Chirdo, F. Structural conformation and self-assembly process of p31-43 gliadin peptide in aqueous solution. Implications for celiac disease. FEBS J. 2019, 287, 2134–2149. [Google Scholar] [CrossRef]
- Castro, M.F.G.; Miculán, E.; Herrera, M.G.; Ruera, C.; Perez, F.; Prieto, E.D.; Barrera, E.; Pantano, S.; Carasi, P.; Chirdo, F.G. p31-43 Gliadin Peptide Forms Oligomers and Induces NLRP3 Inflammasome/Caspase 1- Dependent Mucosal Damage in Small Intestine. Front. Immunol. 2019, 10, 31. [Google Scholar] [CrossRef]
- Bascuñán, K.A.; Araya, M.; Roncoroni, L.; Doneda, L.; Elli, L. Dietary Gluten as a Conditioning Factor of the Gut Microbiota in Celiac Disease. Adv. Nutr. 2019, 11, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Akobeng, A.K.; Singh, P.; Kumar, M.; Al Khodor, S. Role of the gut microbiota in the pathogenesis of coeliac disease and potential therapeutic implications. Eur. J. Nutr. 2020, 59, 3369–3390. [Google Scholar] [CrossRef]
- Polo, A.; Arora, K.; Ameur, H.; Di Cagno, R.; De Angelis, M.; Gobbetti, M. Gluten-free diet and gut microbiome. J. Cereal Sci. 2020, 95, 103058. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Weitnauer, G.; Mühlenweg, A.; Trefzer, A.; Hoffmeister, D.; Süßmuth, R.; Jung, G.; Welzel, K.; Vente, A.; Girreser, U.; Bechthold, A. Biosynthesis of the orthosomycin antibiotic avilamycin A: Deductions from the molecular analysis of the avi biosynthetic gene cluster of Streptomyces viridochromogenes Tü57 and production of new antibiotics. Chem. Biol. 2001, 8, 569–581. [Google Scholar] [CrossRef]
- Viola, R.E. The Central Enzymes of the Aspartate Family of Amino Acid Biosynthesis. Accounts Chem. Res. 2001, 34, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Cargill, J.S.; Scott, K.S.; Gascoyne-Binzi, D.; Sandoe, J.A.T. Granulicatella infection: Diagnosis and management. J. Med. Microbiol. 2012, 61, 755–761. [Google Scholar] [CrossRef]
- Brooks, L.R.K.; Mias, G.I. Streptococcus pneumoniae’s Virulence and Host Immunity: Aging, Diagnostics, and Prevention. Front. Immunol. 2018, 9, 1366. [Google Scholar] [CrossRef]
- Nowruzi, B.; Khavari-Nejad, R.-A.; Sivonen, K.; Kazemi, B.; Najafi, F.; Nejadsattari, T. Identification and toxigenic potential of a Nostoc sp. ALGAE 2012, 27, 303–313. [Google Scholar] [CrossRef]
- McNally, A.; Ross, C.; Wayte, J. Shiitake dermatitis: The tale of an under-recognised, undercooked fungus. Med. J. Aust. 2016, 204, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Urade, R.; Sato, N.; Sugiyama, M. Gliadins from wheat grain: An overview, from primary structure to nanostructures of aggregates. Biophys. Rev. 2018, 10, 435–443. [Google Scholar] [CrossRef]
- Hausch, F.; Shan, L.; Santiago, N.A.; Gray, G.M.; Khosla, C. Intestinal digestive resistance of immunodominant gliadin peptides. Am. J. Physiol. Liver Physiol. 2002, 283, G996–G1003. [Google Scholar] [CrossRef]
- Anderson, O.D.; Dong, L.; Huo, N.; Gu, Y.Q. A New Class of Wheat Gliadin Genes and Proteins. PLoS ONE 2012, 7, e52139. [Google Scholar] [CrossRef]
- Misra, H.S.; Rajpurohit, Y.S.; Khairnar, N.P. Pyrroloquinoline-quinone and its versatile roles in biological processes. J. Biosci. 2012, 37, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.B.; Chowanadisai, W.; Mishchuk, D.O.; Satre, M.A.; Slupsky, C.M.; Rucker, R.B. Dietary pyrroloquinoline quinone (PQQ) alters indicators of inflammation and mitochondrial-related metabolism in human subjects. J. Nutr. Biochem. 2013, 24, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- Bae, T.; Schneewind, O. The YSIRK-G/S Motif of Staphylococcal Protein A and Its Role in Efficiency of Signal Peptide Processing. J. Bacteriol. 2003, 185, 2910–2919. [Google Scholar] [CrossRef]
- Spirig, T.; Weiner, E.M.; Clubb, R.T. Sortase enzymes in Gram-positive bacteria. Mol. Microbiol. 2011, 82, 1044–1059. [Google Scholar] [CrossRef]
- Schneewind, O.; Missiakas, D. Sortases, Surface Proteins, and Their Roles in Staphylococcus aureus Disease and Vaccine Development. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Cossart, P.; Jonquières, R. Sortase, a universal target for therapeutic agents against Gram-positive bacteria? Proc. Natl. Acad. Sci. USA 2000, 97, 5013–5015. [Google Scholar] [CrossRef]
- Gosink, K.K.; Mann, E.R.; Guglielmo, C.; Tuomanen, E.I.; Masure, H.R. Role of Novel Choline Binding Proteins in Virulence of Streptococcus pneumoniae. Infect. Immun. 2000, 68, 5690–5695. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Read, R.; Zhang, Q.; Finn, A. Choline-Binding Protein A ofStreptococcus pneumoniaeElicits Chemokine Production and Expression of Intercellular Adhesion Molecule 1 (CD54) by Human Alveolar Epithelial Cells. J. Infect. Dis. 2002, 186, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Jelínková, L.; Tučková, L.; Cinová, J.; Flegelová, Z.; Tlaskalová-Hogenová, H. Gliadin stimulates human monocytes to production of IL-8 and TNF-α through a mechanism involving NF-κB. FEBS Lett. 2004, 571, 81–85. [Google Scholar] [CrossRef]
- Jelínková, L.; Tučková, L.; Sánchez, D.; Krupičková, S.; Pozler, O.; Nevoral, J.; Kotalová, R.; Tlaskalová-Hogenová, H. Increased levels of circulating ICAM-1, E-selectin, and IL-2 receptors in celiac disease. Dig. Dis. Sci. 2000, 45, 398–402. [Google Scholar] [CrossRef]
- Abel, M.; Cellier, C.; Kumar, N.; Cerf-Bensussan, N.; Schmitz, J.; Caillat-Zucman, S. Adulthood-Onset Celiac Disease Is Associated with Intercellular Adhesion Molecule-1 (ICAM-1) Gene Polymorphism. Hum. Immunol. 2006, 67, 612–617. [Google Scholar] [CrossRef]
- Hammerschmidt, S.; Talay, S.R.; Brandtzaeg, P.; Chhatwal, G.S. SpsA, a novel pneumococcal surface protein with specific binding to secretory Immunoglobulin A and secretory component. Mol. Microbiol. 1997, 25, 1113–1124. [Google Scholar] [CrossRef]
- Zhang, J.-R.; E Mostov, K.; E Lamm, M.; Nanno, M.; Shimida, S.-I.; Ohwaki, M.; Tuomanen, E. The Polymeric Immunoglobulin Receptor Translocates Pneumococci across Human Nasopharyngeal Epithelial Cells. Cell 2000, 102, 827–837. [Google Scholar] [CrossRef]
- Maestro, B.; Sanz, J.M. Choline Binding Proteins from Streptococcus pneumoniae: A Dual Role as Enzybiotics and Targets for the Design of New Antimicrobials. Antibiotics 2016, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Matysiak-Budnik, T.; Moura, I.C.; Arcos-Fajardo, M.; Lebreton, C.; Menard, S.; Candalh, C.; Ben-Khalifa, K.; Dugave, C.; Tamouza, H.; Van Niel, G.; et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 2007, 205, 143–154. [Google Scholar] [CrossRef]
- Murakami, S. Multidrug efflux transporter, AcrB—the pumping mechanism. Curr. Opin. Struct. Biol. 2008, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Ramm, B.; Heermann, T.; Schwille, P. The E. coli MinCDE system in the regulation of protein patterns and gradients. Cell. Mol. Life Sci. 2019, 76, 4245–4273. [Google Scholar] [CrossRef]
- Koonin, E.V. A Superfamily of ATPases with Diverse Functions Containing Either Classical or Deviant ATP-binding Motif. J. Mol. Biol. 1993, 229, 1165–1174. [Google Scholar] [CrossRef]
- Vecchiarelli, A.G.; Mizuuchi, K.; Funnell, B.E. Surfing biological surfaces: Exploiting the nucleoid for partition and transport in bacteria. Mol. Microbiol. 2012, 86, 513–523. [Google Scholar] [CrossRef]
- Hester, C.M.; Lutkenhaus, J. Soj (ParA) DNA binding is mediated by conserved arginines and is essential for plasmid segregation. Proc. Natl. Acad. Sci. USA 2007, 104, 20326–20331. [Google Scholar] [CrossRef] [PubMed]
- Castaing, J.-P.; Bouet, J.-Y.; Lane, D. F plasmid partition depends on interaction of SopA with non-specific DNA. Mol. Microbiol. 2008, 70, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Soberón, N.E.; Lioy, V.; Pratto, F.; Volante, A.; Alonso, J.C. Molecular anatomy of the Streptococcus pyogenes pSM19035 partition and segrosome complexes. Nucleic Acids Res. 2010, 39, 2624–2637. [Google Scholar] [CrossRef]
- Hayashi, I.; Oyama, T.; Morikawa, K. Structural and functional studies of MinD ATPase: Implications for the molecular recognition of the bacterial cell division apparatus. EMBO J. 2001, 20, 1819–1828. [Google Scholar] [CrossRef]
- Zhou, H.; Lutkenhaus, J. MinC Mutants Deficient in MinD- and DicB-Mediated Cell Division Inhibition Due to Loss of Interaction with MinD, DicB, or a Septal Component. J. Bacteriol. 2005, 187, 2846–2857. [Google Scholar] [CrossRef]
- Lutkenhaus, J. The ParA/MinD family puts things in their place. Trends Microbiol. 2012, 20, 411–418. [Google Scholar] [CrossRef]
- Oldstone, M.B.A. Molecular mimicry and immune-mediated diseases. FASEB J. 1998, 12, 1255–1265. [Google Scholar] [CrossRef]
- Kohm, A.P.; Fuller, K.G.; Miller, S.D. Mimicking the way to autoimmunity: An evolving theory of sequence and structural homology. Trends Microbiol. 2003, 11, 101–105. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.-M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Rial, J.; Calle, I.R.; Salas, A.; Martinón-Torres, F. Rotavirus and autoimmunity. J. Infect. 2020, 81, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Cuan-Baltazar, Y.; Soto-Vega, E. Microorganisms associated to thyroid autoimmunity. Autoimmun. Rev. 2020, 19, 102614. [Google Scholar] [CrossRef] [PubMed]
- Múnera, M.; Farak, J.; Pérez, M.; Rojas, J.; Villero, J.; Sánchez, A.; Emiliani, Y. Prediction of molecular mimicry between antigens from Leishmania sp. and human: Implications for autoimmune response in systemic lupus erythematosus. Microb. Pathog. 2020, 148, 104444. [Google Scholar] [CrossRef]
- Cunningham, M.W. Streptococcus and rheumatic fever. Curr. Opin. Rheumatol. 2012, 24, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.W. Molecular Mimicry, Autoimmunity, and Infection: The Cross-Reactive Antigens of Group A Streptococci and their Sequelae. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Lucchese, G.; Flöel, A. SARS-CoV-2 and Guillain-Barré syndrome: Molecular mimicry with human heat shock proteins as potential pathogenic mechanism. Cell Stress Chaperones 2020, 25, 731–735. [Google Scholar] [CrossRef]
- Ozuna, C.V.; Iehisa, J.C.M.; Gimenez, M.J.; Alvarez, J.B.; Sousa, C.; Barro, F. Diversification of the celiac disease α-gliadin complex in wheat: A 33-mer peptide with six overlapping epitopes, evolved following polyploidization. Plant J. 2015, 82, 794–805. [Google Scholar] [CrossRef]
- Molberg, Ø.; Mcadam, S.N.; Körner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Norén, O.; Roepstorff, P.; et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Arentz-Hansen, H.; Körner, R.; Molberg, Ø.; Quarsten, H.; Vader, W.; Kooy, Y.M.; Lundin, K.E.; Koning, F.; Roepstorff, P.; Sollid, L.M.; et al. The Intestinal T Cell Response to α-Gliadin in Adult Celiac Disease Is Focused on a Single Deamidated Glutamine Targeted by Tissue Transglutaminase. J. Exp. Med. 2000, 191, 603–612. [Google Scholar] [CrossRef]
- Qiao, S.-W.; Bergseng, E.; Molberg, Ø.; Jung, G.; Fleckenstein, B.; Sollid, L.M. Refining the Rules of Gliadin T Cell Epitope Binding to the Disease-Associated DQ2 Molecule in Celiac Disease: Importance of Proline Spacing and Glutamine Deamidation. J. Immunol. 2005, 175, 254–261. [Google Scholar] [CrossRef]
- Ruiz-Carnicer, Á.; Comino, I.; Segura, V.; Ozuna, C.V.; Moreno, M.D.L.; López-Casado, M.Á.; Torres, M.I.; Barro, F.; Sousa, C. Celiac Immunogenic Potential of α-Gliadin Epitope Variants from Triticum and Aegilops Species. Nutrients 2019, 11, 220. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein–protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef]
- Teyra, J.; Huang, H.; Jain, S.; Guan, X.; Dong, A.; Liu, Y.; Tempel, W.; Min, J.; Tong, Y.; Kim, P.M.; et al. Comprehensive Analysis of the Human SH3 Domain Family Reveals a Wide Variety of Non-canonical Specificities. Struct. 2017, 25, 1598–1610.e3. [Google Scholar] [CrossRef]
- Chen, H.I.; Sudol, M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Ball, L.J.; Kühne, R.; Schneider-Mergener, J.; Oschkinat, H. Recognition of Proline-Rich Motifs by Protein-Protein-Interaction Domains. Angew. Chem. Int. Ed. 2005, 44, 2852–2869. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Sudol, M. The WW domain: A signalling site in dystrophin? Trends Biochem. Sci. 1994, 19, 531–533. [Google Scholar] [CrossRef]
- Méthot, P.-O.; Alizon, S. What is a pathogen? Toward a process view of host-parasite interactions. Virulence 2014, 5, 775–785. [Google Scholar] [CrossRef]
- Ingram, J.H.; Stone, M.; Fisher, J.; Ingham, E. The influence of molecular weight, crosslinking and counterface roughness on TNF-alpha production by macrophages in response to ultra high molecular weight polyethylene particles. Biomaterials 2004, 25, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.; Green, T.R.; Stone, M.H.; Wroblewski, B.M.; Fisher, J.; Ingham, E. Comparison of the response of primary human peripheral blood mononuclear phagocytes from different donors to challenge with model polyethylene particles of known size and dose. Biomaterials 2000, 21, 2033–2044. [Google Scholar] [CrossRef]
- Green, T. Polyethylene particles of a ‘critical size’ are necessary for the induction of cytokines by macrophages in vitro. Biomaterials 1998, 19, 2297–2302. [Google Scholar] [CrossRef]
- Paul, D.; Achouri, S.; Yoon, Y.-Z.; Herre, J.; Bryant, C.E.; Cicuta, P. Phagocytosis Dynamics Depends on Target Shape. Biophys. J. 2013, 105, 1143–1150. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Pokhrel, A.R.; Nguyen, C.T.; Pham, V.T.T.; Dhakal, D.; Lim, H.N.; Jung, H.J.; Kim, T.-S.; Yamaguchi, T.; Sohng, J.K. Streptomyces sp. VN1, a producer of diverse metabolites including non-natural furan-type anticancer compound. Sci. Rep. 2020, 10, 1756. [Google Scholar] [CrossRef]
- Ebadi, M.; Zolfaghari, M.R.; Aghaei, S.S.; Zargar, M.; Shafiei, M.; Zahiri, H.S.; Noghabi, K.A. A bio-inspired strategy for the synthesis of zinc oxide nanoparticles (ZnO NPs) using the cell extract of cyanobacterium Nostoc sp. EA03: From biological function to toxicity evaluation. RSC Adv. 2019, 9, 23508–23525. [Google Scholar] [CrossRef]
- Sanchez, C.J.; Kumar, N.; Lizcano, A.; Shivshankar, P.; Hotopp, J.C.D.; Jorgensen, J.H.; Tettelin, H.; Orihuela, C.J. Streptococcus pneumoniae in Biofilms Are Unable to Cause Invasive Disease Due to Altered Virulence Determinant Production. PLoS ONE 2011, 6, e28738. [Google Scholar] [CrossRef]
- Karched, M.; Bhardwaj, R.G.; Asikainen, S.E. Coaggregation and biofilm growth of Granulicatella spp. with Fusobacterium nucleatum and Aggregatibacter actinomycetemcomitans. BMC Microbiol. 2015, 15, 114. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, G.; Fahlgren, A.; Horstedt, P.; Hammarstrom, S.; Hernell, O.; Hammarstrom, M.-L. Presence of Bacteria and Innate Immunity of Intestinal Epithelium in Childhood Celiac Disease. Am. J. Gastroenterol. 2004, 99, 894–904. [Google Scholar] [CrossRef]
- Corazza, G.R.; Strocchi, A.; Gasbarrini, G. Fasting breath hydrogen in celiac disease. Gastroenterology 1987, 93, 53–58. [Google Scholar] [CrossRef]
- Tursi, A.; Brandimarte, G.; Giorgetti, G. High prevalence of small intestinal bacterial overgrowth in celiac patients with persistence of gastrointestinal symptoms after gluten withdrawal. Am. J. Gastroenterol. 2003, 98, 839–843. [Google Scholar] [CrossRef]
- Valitutti, F.; Cucchiara, S.; Fasano, A. Celiac Disease and the Microbiome. Nutrients 2019, 11, 2403. [Google Scholar] [CrossRef]
- Pascual, V. Inflammatory bowel disease and celiac disease: Overlaps and differences. World J. Gastroenterol. 2014, 20, 4846–4856. [Google Scholar] [CrossRef]
- Francavilla, R.; Ercolini, D.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Filippis, F.; De Pasquale, I.; Di Cagno, R.; Di Toma, M.; Gozzi, G.; et al. Salivary Microbiota and Metabolome Associated with Celiac Disease. Appl. Environ. Microbiol. 2014, 80, 3416–3425. [Google Scholar] [CrossRef] [PubMed]
- Olivares, M.; Neef, A.; Castillejo, G.; De Palma, G.; Varea, V.; Capilla, A.; Palau, F.; Nova, E.; Marcos, A.; Polanco, I.; et al. The HLA-DQ2 genotype selects for early intestinal microbiota composition in infants at high risk of developing coeliac disease. Gut 2015, 64, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Ou, G.; Hedberg, M.; Hörstedt, P.; Baranov, V.; Forsberg, G.; Drobni, M.; Sandström, O.; Wai, S.N.; Johansson, I.; Hammarström, M.-L.; et al. Proximal Small Intestinal Microbiota and Identification of Rod-Shaped Bacteria Associated with Childhood Celiac Disease. Am. J. Gastroenterol. 2009, 104, 3058–3067. [Google Scholar] [CrossRef] [PubMed]
- Segal, S.; Hill, A.V. Genetic susceptibility to infectious disease. Trends Microbiol. 2003, 11, 445–448. [Google Scholar] [CrossRef]
- Zhou, B.; Yuan, Y.; Zhang, S.; Guo, C.; Li, X.; Li, G.; Xiong, W.; Zeng, Z. Intestinal Flora and Disease Mutually Shape the Regional Immune System in the Intestinal Tract. Front. Immunol. 2020, 11, 575. [Google Scholar] [CrossRef] [PubMed]
- Stepniak, D.; Koning, F. Celiac Disease—Sandwiched between Innate and Adaptive Immunity. Hum. Immunol. 2006, 67, 460–468. [Google Scholar] [CrossRef]
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori Infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Case, D.A. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Knapp, B.; Lederer, N.; Omasits, U.; Schreiner, W. vmdICE: A plug-in for rapid evaluation of molecular dynamics simulations using VMD. J. Comput. Chem. 2010, 31, 2868–2873. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zambrano, R.; Jamroz, M.; Szczasiuk, A.; Pujols, J.; Kmiecik, S.; Ventura, S. AGGRESCAN3D (A3D): Server for prediction of aggregation properties of protein structures. Nucleic Acids Res. 2015, 43, W306–W313. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name * (Reference) | Organism | Sequence Identity ** (E-Value) | Sequence Similarity (Subcellular and Topological Localization ***) | Organism Pathogenicity in Humans |
|---|---|---|---|---|
| BLASTp search using the 33mer sequence as query (A) | ||||
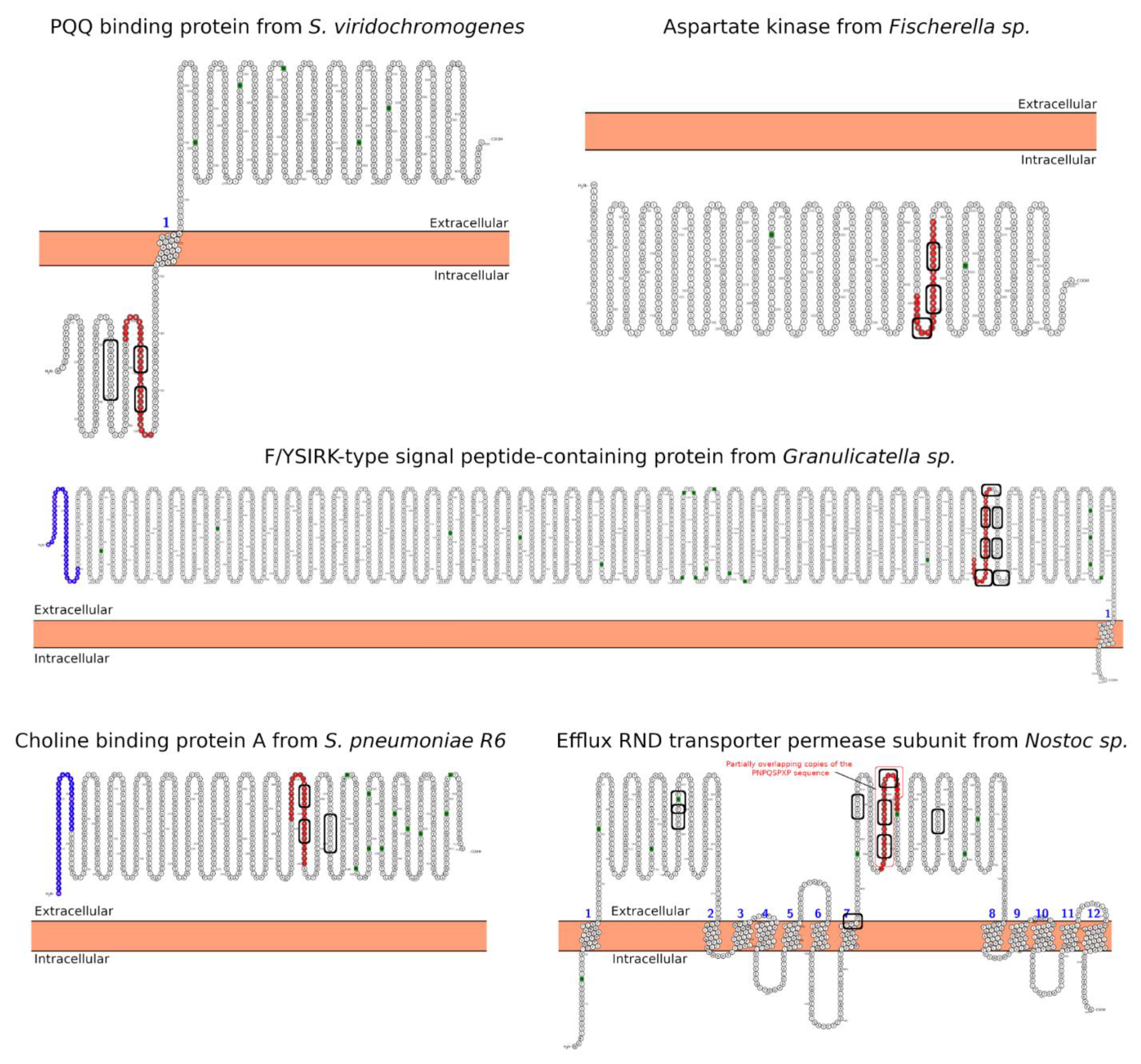
| Hit#1: PQQ-repeat protein (NCBI: WP_048584697.1) | Streptomyces viridochromogenes | 68% (0.004) | 95PYGQQQDPYGQPQAPYGQPQAPYGQPQP122 (T(s-p)/I) | Not reported; producer of bioactive compounds, e.g., antibiotics [31]. |
| Hit#2: Aspartate kinase (NCBI: WP_017312730.1) | Fischerella sp. | 54% (0.064) | 459PIPNPQSPIPNPQSPIPDPRSPIPDP484 (I) | Not reported; producer of secondary metabolites [32]. |
| Hit#3: F/YSIRK-type signal peptide-containing protein (NCBI: WP_070445895.1) | Granulicatella sp. HMSC31F03 | 46% (0.075) | 1747QPNPDPEKPTPDPEKPTPDPEKPTPDPE1774 (T(s-p)/E) | Potentially pathogenic [33]; commensal of mucosal surfaces. |
| Hit#4: Choline-binding protein A (NCBI: WP_000458116.1) | Streptococcus pneumoniae R6 | 45% (0.079) | 412KPAEQPQPAPATQPEKPAPKPEKPAEQPK440 (E) | Opportunistic pathogen [34]; mucosal surface in upper respiratory tract-forming biofilms. |
| Hit#5: Efflux RND transporter permease subunit (NCBI: WP_015136936.1) | Nostoc sp. | 54% (0.097) | 535LPNPQSPIPNPQSPVPNPQSPIPINPL562 (T(m-p)/E) | Potentially low pathogenic via ecotoxicology [35]; producer of toxic compounds and bioactive compounds with pharmaceutical potential. |
| BLASTp search using the p31-43 sequence as query (B) | ||||
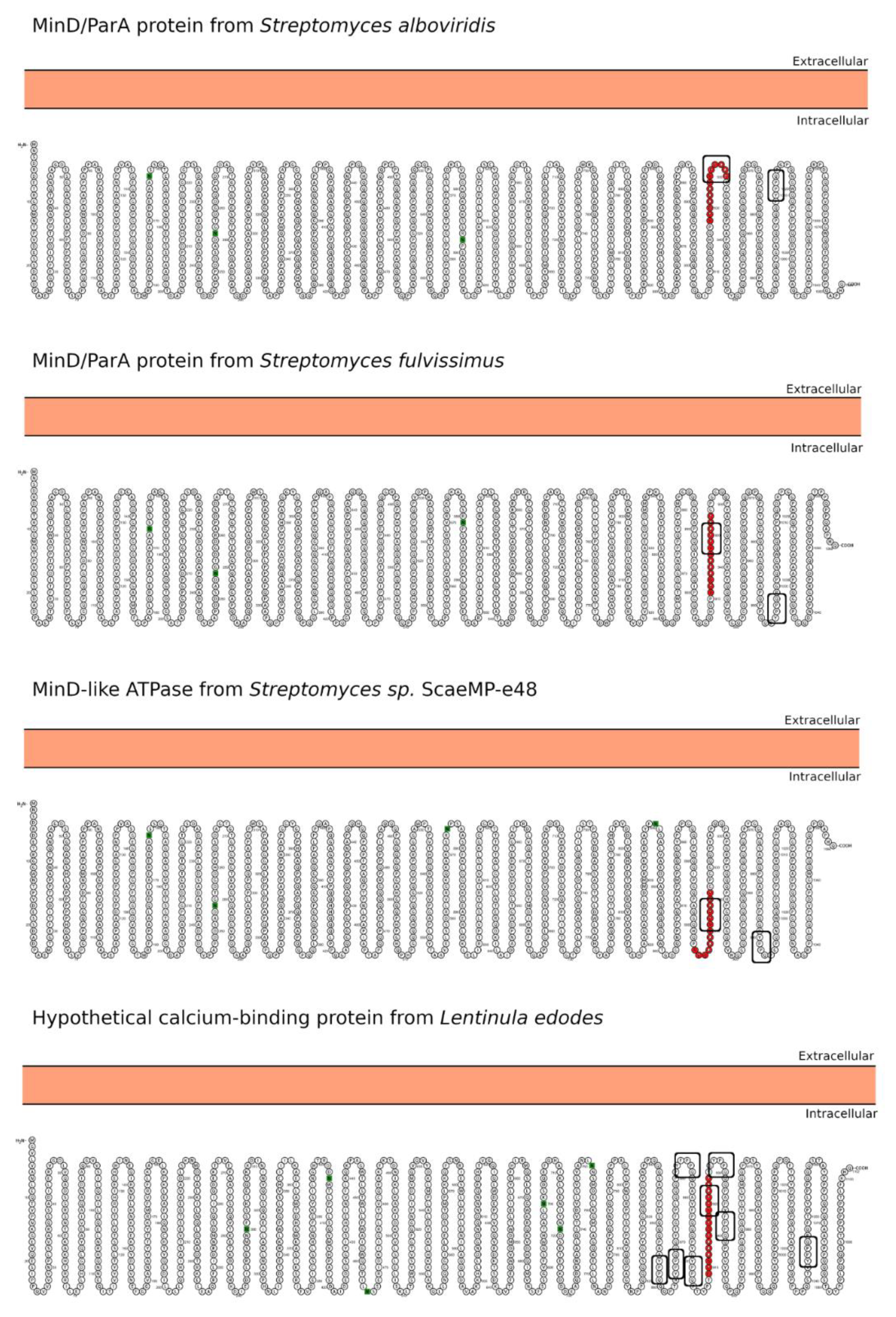
| Hit#1: MinD/ParA family protein (NCBI: WP_032757377.1) | Streptomyces alboviridis | 85% (0.003) | 918YPGQQQPYPPQQP930 (I) | Not reported. |
| Hit#2: MinD/ParA family protein (NCBI: WP_015611830.1) | Streptomyces fulvissimus | 85% (0.003) | 911YPGQQQPYPPQQP923 (I) | Not reported. |
| Hit#3: MinD-like ATPase (GenBank: SCK04981.1) | Streptomyces sp. ScaeMP-e48 | 85% (0.003) | 904YPGQQQPYPPQQP916 (I) | Not reported. |
| Hit#4: Hypothetical calcium-binding protein LENED-006428 (GenBank: GAW04623.1) | Lentinula edodes | 75% (0.024) | 909FPSQQQPAPYFPPQP924 (I) | Potentially pathogenic [36] causing, e.g., dermatitis herpetiformis; mushroom used in traditional medicine. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vazquez, D.S.; Schilbert, H.M.; Dodero, V.I. Molecular and Structural Parallels between Gluten Pathogenic Peptides and Bacterial-Derived Proteins by Bioinformatics Analysis. Int. J. Mol. Sci. 2021, 22, 9278. https://doi.org/10.3390/ijms22179278
Vazquez DS, Schilbert HM, Dodero VI. Molecular and Structural Parallels between Gluten Pathogenic Peptides and Bacterial-Derived Proteins by Bioinformatics Analysis. International Journal of Molecular Sciences. 2021; 22(17):9278. https://doi.org/10.3390/ijms22179278
Chicago/Turabian StyleVazquez, Diego S., Hanna M. Schilbert, and Veronica I. Dodero. 2021. "Molecular and Structural Parallels between Gluten Pathogenic Peptides and Bacterial-Derived Proteins by Bioinformatics Analysis" International Journal of Molecular Sciences 22, no. 17: 9278. https://doi.org/10.3390/ijms22179278